Among many papers on Geometric and Graph ML, applications in biochemistry, drug design, and structural biology shone in 2020. Perhaps for the first time, we are finally starting to see the real impact of these machine learning techniques in fundamental science. In this post, I highlight three papers that excited me the most in the past year (disclaimer: I am a co-author of one of them).
J. M. Stokes et al., A deep learning approach to antibiotic discovery (2020) Cell 180(4):688–702.
What? A graph neural network-based deep learning pipeline for the discovery of new antibiotic drugs.
How? A graph neural network was trained to predict the growth inhibition of the bacterium Escherichia coli on a dataset of >2000 molecules (including approved antibiotic drugs as well as natural compounds from the plant and animal kingdoms) with known antibacterial activity. The prediction is based on the molecular graph only and does not rely on any side information such as the drug mechanism of action.
The trained model was then applied to the Drug Repurposing Hub, containing around 6000 molecules of drugs under investigation. The top 100 candidates were selected for lab testing. Surprisingly, an experimental anti-diabetic drug halicin turned out to be a potent antibiotic that showed activity against multiple drug-resistant pathogens in test mice. The graph neural network apparently has good generalisation capabilities, as the halicin molecule does not resemble traditional antibiotics. Yet, it is not completely clear whether its predictive capability boils down to predicting a simple pattern responsible for the antibiotic action hinted in the paper (depolarisation of the cellular membrane).
Additional experiments were carried out to screen more than 100 million molecular structures in the ZINC15 database, a curated collection of commercially available chemical compounds prepared especially for virtual screening and routinely used by drug designers. Among the shortlisted compounds, physical tests identified 8 with antibacterial activity, of which two were shown to have potent activity against a broad range of pathogens.
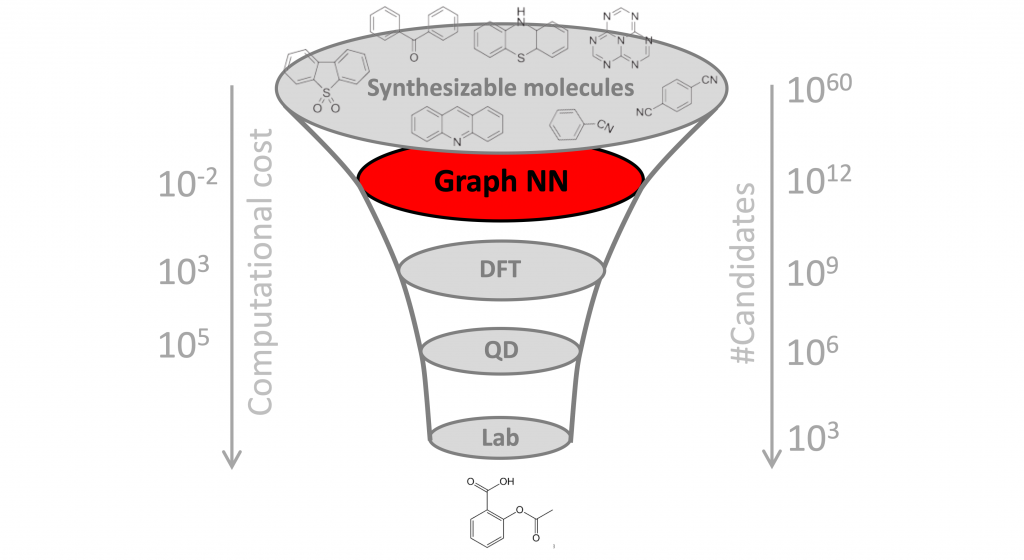
Why it matters? One of the key challenges in drug discovery is the vast search space, estimated to contain over 10⁶⁰ molecules. Since only a tiny fraction of these molecules can be tested in the lab, it is paramount to select promising candidates. Doing this computationally is called “virtual screening”. While ML methods have been used in the past for virtual screening of molecules and, more broadly, to assist different stages of drug development and discovery, this is the first time a completely new class of antibiotics has been identified from scratch, without using any previous human assumptions.
Unlike the majority of in silico ML-based drug discovery papers that end with only a computational prediction, the paper of Stokes et al. not only identified promising molecules but also extensively validated them in vivo on lab animals. While the approach can in principle be applied to discover candidate therapies against other diseases such as cancer, the focus on antibiotics is very timely: antibiotic-resistant microorganisms that can develop as a result of antibiotic abuse are one of the nightmares of world-wide healthcare, and the potential emergence of a highly-contagious bacterial infection that evades the existing arsenal of drugs is not a question of “if” but “when”.
More on this: Quanta Magazine popular article and Jim Collins’ TED Talk 2020 (Collins lab was part of this year’s TED Audacious Projects cohort, together with our Project CETI).
J. Jumper et al., High accuracy protein structure prediction using deep learning (2020) a.k.a. AlphaFold 2.0(paper not yet available)
What? Very accurate prediction of 3D protein structure from aminoacid sequence, a notoriously hard problem in bioinformatics.
How? AlphaFold 2.0 is an “attention-based neural network” (likely a transformer architecture) trained end-to-end on 170000 protein structures from the Protein Data Bank as well as protein sequences with unknown structure. Since DeepMind has not yet disclosed the details of the algorithm, we can only hypothesise how it works. From the blog post, proteins are modelled as spatial contact graphs, and the neural network “interprets the structure of this graph, while reasoning over the implicit graph that it is building.” It sounds like some form of graph neural network with latent graph learning, though there are probably many more additional details and nuances, as the method also uses evolutionarily sequence information, so I would classify it as “geometric ML” with some caution. The reported computational complexity of training is very high (an equivalent of several years of GPU time) and the prediction of the structure is “a matter of days.”

Why it matters? Proteins are arguably the most important biomolecules, often called the “molecules of life”, as we are currently not aware of any life-form that is not protein-based. Proteins are encoded in our DNA and have myriads of functions in our body, including protection against pathogens (antibodies), giving structure to our skin (collagen), transporting oxygen to cells (haemoglobin), catalysing chemical reactions (enzymes), and signaling (many hormones are proteins).
Chemically speaking, a protein is a biopolymer, or a chain of small building blocks called aminoacids that under the influence of electrostatic forces fold into a complex 3D structure. It is this structure that endows the protein with its functions, and hence it is crucial to the understanding of how proteins work and what they do. Since proteins are common targets for drug therapies (typical drugs are small molecules designed to bind to their target), the pharmaceutical industry has a keen interest in this field.
Modern technology allows to sequence proteins (i.e. obtain the string of their constituent aminoacids) cheaply and reliably but getting the 3D structure typically relies on old crystallographic techniques that are capricious, long, and expensive. As a result, there are about 200 million proteins with known sequences and less than 200 thousand with known structures.
It was long hypothesised that the aminoacid sequence contains enough information to predict the 3D structure of the protein, but the problem turned to be extremely challenging. The Critical Assessment of Protein Structure Prediction (CASP), an ImageNet-like competition running since 1994 in which participants try to predict 3D structures of unknown proteins, has been a classical testbed for bioinformatics research labs and pharmaceutical companies.
In 2018, DeepMind surprised the research community with a newcomer AlphaFold that came out of the blue as the winner of CASP. The 2020 version of the algorithm, AlphaFold 2.0 seems to work significantly better, achieving a root mean squared distance (RMSD) of 1.6 angstroms, which is considered very accurate by structural biology standards and a far cry from other competitors. This is an “ImageNet moment” for protein science.
While by all means stunning progress on a key problem, the popular press hype with relaxed terminology provides a somewhat distorted picture of what AlphaFold can or cannot do. In particular, for drug design applications, one typically requires sub-angstrom accuracy on the binding site that the method cannot yet deliver.
More on this: Everyone is eagerly waiting for the paper detailing the algorithm to be published. In the meantime, Lex Fridman’s YouTube video provides a good summary, and Mohammed AlQuraishi’s blog post describes the effect AlphaFold has had in 2018.
P. Gainza et al., Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning (2020) Nature Methods 17(2):184–192.
What? A geometric deep learning pipeline called MaSIF predicting interactions between proteins from their 3D structure.
How? MaSIF models the protein as a molecular surface discretised as a mesh, arguing that this representation is advantageous when dealing with interactions as it allows to abstract the internal fold structure. The architecture is based on MoNet, a mesh convolutional neural network developed by my PhD student Federico Monti and operating on pre-computed chemical and geometric features in small geodesic patches.
The network is trained using a few thousand co-crystal protein 3D structures from the Protein Data Bank to address multiple tasks, including interface prediction, ligand classification, and docking, showing state-of-the-art performance. A key difference of MaSIF compared to other methods is that it does not rely on the evolutionary history of proteins. This is of crucial importance in de novo protein design, where one tries to create “from scratch” new proteins that have never existed before.
Allowing myself to be more critical as a co-author of the paper, I would highlight the need to precompute the molecular surface mesh and local patches as well as the reliance on hand-crafted features as one of the key drawbacks of MaSIF. Over the course of this year, we have completely revamped the architecture, which now operates directly on the atomic point cloud as input, computes the molecular surface (represented as a point cloud) on the fly, learns geometric and chemical features, is end-to-end differentiable, and runs orders of magnitude faster (the latter is afforded due to the use of a fast geometric computation library KeOps, developed by my postdoc Jean Feydy). Moreover, while the Nature Methods paper focused primarily on the computational methodology, the crystallographic structures of several of the new protein binders designs with MaSIF were subsequently obtained by my EPFL collaborators, coinciding very accurately with the computed one.
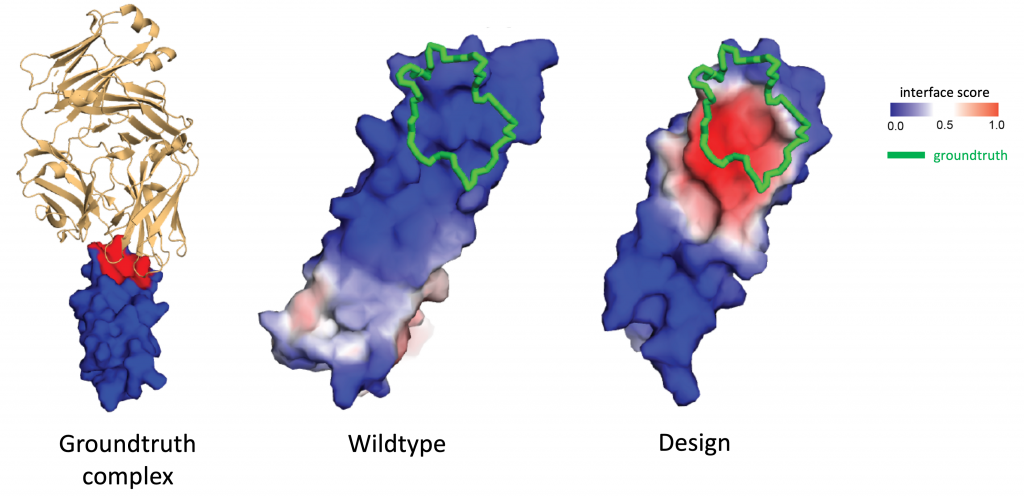
Why it matters? Interactions between proteins and other biomolecules are the basis of protein function in most biological processes. A better understanding of how proteins interact is thus key both for fundamental biology as well as for drug development: many diseases are associated with protein-to-protein interactions (PPI) which are promising drug targets. However, such interactions usually involve flat interfaces deemed “undruggable”, as they are very different from the traditional pocket-shaped structures targeted by small drug molecules.
The success of MaSIF to identify binders for such targets make it a promising tool for rational protein design and opens multiple exciting applications in biological drug development, such as immune checkpoint inhibition cancer therapy that tries to target the PD-1/PD-L1 protein complex responsible for programmed cell death.
More on this: John Pavlus’ article in Quanta Magazine (John has previously reported on geometric deep learning), Bharath Ramsundar’s blog post, and my talk at the Broad Institute earlier this year.
I am grateful to Sergey Ivanov, Maksym Korablyov, and Jake Taylor-King for comments on an early draft of this post.